The term microtia literally means “small ear” but in reality indicates a small, abnormally shaped or absent external ear. Microtia can occur on one side only (unilateral) or on both sides ("bilateral). The unilateral form is much more common, occurring in approximately 90% of patients. As will be discussed below, microtia is tremendously variable. Some patients have small, almost normal appearing ears while others have completely absent ears. Some patients have no evidence of an ear canal and some have a blind canal. While some patients appear to have a true ear canal, these canals are almost always blind and to not communicate with the middle ear (more on this below under Aural Atresia). Some patients have a hairline that is in normal position, leaving appropriate, hairless skin for the reconstructed ear. Other patients have a low hairline and additional maneuvers are required to avoid a reconstructed ear that is partially or completely covered with hair.
The term aural atresia refers to the absence of the ear canal and ear drum. Patients who have microtia usually, but not always, have aural atresia. The reverse can also be true: some patients have aural atresia but a normal appearing external ear. The most common situation, however, is the patient with microtia AND aural atresia. Patients who have aural atresia do not have good hearing on that side but usually have completely normal hearing on the opposite side. Because of the normal hearing on the opposite side, these patients usually have no trouble hearing when someone speaks loudly to them in a quiet surrounding environment or in one-on-one conversations. Patients who hear on only one side, however, have difficulty localizing sounds and have difficulty distinguishing sounds in situations where there is background noise (play-ground, classrooms, parties etc).
It is important to note that patients who have atresia in both ears will be sufficiently hard of hearing to be totally dependent on a hearing aid in all situations (See below)
Patients who lack the ear canal are not just missing a “hole” in the skin; they also have no canal through in the skull. In other words, the outer ear is completely separated from the middle ear by bone. No wonder the sound can’t get through! These patients also have structural abnormalities of the middle ear itself with absence of the eardrum and incomplete formation of the tiny middle ear bones, which normally allow conduction of hearing through the middle ear. Microtia and aural atresia tend to occur together because the outer ear and the middle ear develop together in fetal life. The inner ear, on the other hand, develops with the brain, and is almost always normal in patients with microtia and aural atresia.

Diagram of Normal External Ear
In most patients microtia and aural atresia occur as isolated conditions. In approximately 10% patients, however, the ear deformity occurs in conjunction with other facial abnormalities. The most common condition in which microtia accompanies other anomalies is called "hemifacial microsomia". This condition has several other names which makes it difficult to research for patients and families. For example, hemifacial microsomia is also known as the "1st and 2nd branchial arch syndrome" and the "oto-mandibular syndrome" as well as by other names. This condition is also tremendously variable and involves underdevelopment, to some degree, of all the structures on one side of the face, including the ear, the jaw bones, the fullness of the cheek tissue, function of the facial nerve and the movement of the facial muscles. Like isolated microtia, hemifacial microsomia can also occur, in a minority of patients, on both sides of the face.

A patient with classic microtia and aural atresia (absent ear canal). An Earlobe is present but is in the wrong location.
Another syndrome where microtia may be present is Treacher Collins Syndrome. This condition is always bilateral and tends to be an inherited syndrome and involves underdevelopment of the lower eyelids, the cheekbones, and the lower jaw. Patients who have severe manifestations of the syndrome frequently have upper airway obstruction and require a tracheostomy and/or surgical elongation of the lower jaw at a very young age. Treacher Collins is inherited such that, statistically, half of the children born to a Treacher Collins patient will be affected to some degree. I say “statistical” because a Treacher Collins patient may have three unaffected children and another patient may have three affected children. “Statistically” means that on average, like flipping a coin, half the offspring will be affected.
Because of the association with these other conditions, patients with microtia may require access to other specialists and may require a genetics evaluation so that parents can be informed of any increase risk of these conditions in future children.
In most patients the only issue that requires attention early in childhood is an evaluation of the hearing. A hearing test (called an audiogram) will be recommended shortly after birth. There are two types of hearing tests, BAER testing (Brain Stem Auditory Evoked Response testing) and Behavioral Testing. BAER testing is performed before the child is old enough to cooperate with behavioral testing. BAER testing determines if the child has any hearing, but it does NOT indicate which side the child is hearing from. In other words, unilateral microtia patients will have normal or near normal BAER testing, indicating that hearing is present. Some parents think this means that the hearing is totally normal. This is not true. It simply means that hearing is present on at least one side.
Reliable behavioral testing is the hearing test of choice when the child is mature enough to cooperate. Behavioral testing can accurately evaluate the hearing in each ear and document that the microtic side has a conductive hearing loss and the other side has normal hearing.
As described above, patients who have involvement of one side of the face only (unilateral), almost always have normal hearing in the other ear and will consequently have adequate hearing function in some situations. Unilateral patients may do reasonably well without a hearing aid but can achieve binaural hearing (hearing on both sides), and may function better in the classroom and social situations, by wearing a headband with an attached bone conduction hearing aid known as a BAHA softband.
The 10% of patients in whom the condition affects both ears (bilateral aural atresia), however, are functionally deaf. It is essential that they receive bone conduction hearing aids as early in life as possible. Patients who are deaf and do not receive hearing aids will not have sufficient hearing to develop normal speech.
To reiterate, patients with a unilateral deformity usually have normal hearing in the opposite ear. They have functional hearing but will have difficulty localizing sounds and in discriminating sound in situations where there is background noise. These patients benefit from bone conduction hearing aids mentioned above. In a minority of patients the condition affects both ears and these patients will absolutely require a hearing aid in order to function in society and develop adequate speech.
The bone conduction hearing aids will be recommended within the first few months of life. Later in life it will be determined if the child is best served by a Bone Anchored Hearing Aid (BAHA) or by surgical reconstruction of the hearing. Surgical reconstruction is only possible in some patients and depends on how abnormal the middle ear anatomy is. The recommendation for a fixed BAHA or surgical reconstruction is not made for several years. The skull must be thick enough for the BAHA implant and, in the case of surgical reconstruction, the patient must have already had the outer ear reconstructed.
Treacher Collins patients usually have sufficient anatomic abnormalities of the middle ear that the hearing cannot be surgically reconstructed and these patients are usually dependent on a hearing aid for life.
Regular monitoring of hearing and language is important and should be performed annually and after every ear infection. Since patients who have microtia/aural atresia on one side are completely dependent on the other ear for hearing, it is important to make sure that the hearing remains normal in that other ear. Middle ear infections (otitis media) can result in accumulation of fluid in the middle ear, which can significantly reduce hearing. You can imagine the situation where a patient with unilateral microtia/aural atresia on the right side, develops fluid accumulation in the left ear; that patient’s hearing would be significantly compromised. Middle ear fluid is easily treatable and therefore regular follow up by a pediatric otolaryngologist is essential before speech is affected.
A high resolution CT scan will indicate if the middle ear structures can be surgically reconstructed. In other words, all patients with microtia are candidates for surgical reconstruction of the outer ear (auricle), but not all patients with aural atresia are candidates for reconstruction of the middle ear and ear canal – it depends on how abnormal or underdeveloped the middle ear structures are.
Patients with aural atresia on one side have functional hearing in some situations. These patients will experience difficulty localizing and discriminating sounds because they lack the stereo effect of having two ears. Unilateral patients should be evaluated by an experienced Otologist regarding the possibility of a BAHA or surgical reconstruction of a canal and the middle ear. Dr. Thorne works with Otologists in many cases. The surgical reconstruction of the hearing has improved dramatically over recent years and some families opt to have the middle ear reconstruction, when possible, on the affected side. This is an individual patient/family decision, which balances the benefit of having "stereo" hearing vs. having an additional procedure. In addition, the improvement in hearing that can be dramatic immediately post operatively, tends to deteriorate to some degree with time. The surgery to reconstruct the middle ear also adversely affects the aesthetics of the outer ear reconstruction to some degree.
This is an important question, which has undergone evolution. Up until ten years ago, surgical reconstruction of the outer ear was recommended beginning at the age of approximately six years. At this age the patients were thought to have sufficient cartilage in the rib cage to allow reconstruction of the ear. As surgical techniques have improved, however, it is clear that a better quality, more detailed, ear reconstruction is possible when the surgery is delayed to after the age of 10 years. Parents all want the same thing: what is best for their child. When first hearing that reconstruction is not recommended for a few additional years, parents are inevitably disappointed. We have never seen, however, a child suffer psychological distress because of delaying the reconstruction. Remember, the child will have this ear for about 90 years, hopefully, and we want it to be as ideal as possible.
If surgery is recommended for reconstruction of the middle ear, it only takes place after the outer ear has been reconstructed. It is important that the outer ear and middle ear reconstructions be coordinated. If the middle ear procedure is performed first, it may eliminate the chances for reconstruction of the outer ear.
Ear reconstruction is generally accomplished in two stages, separated by approximately 6 months. Occasionally additional “touch up” procedures are performed in order to maximize the aesthetics of the final result.
It is often difficult for parents to deal with their own responses to their child's condition as well as the responses of other people. It may be helpful to speak to other families who have children with similar conditions.
Variability of microtia. The images below demonstrate the vast difference in the appearance of microtia. The reconstructive technique must be customized for each patient.

Large conchal microtia

Small conchal microtia
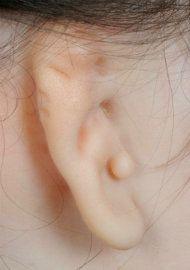
Lobular microtia
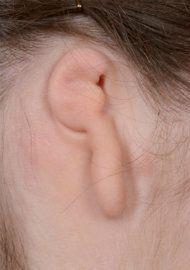
Lobular microtia
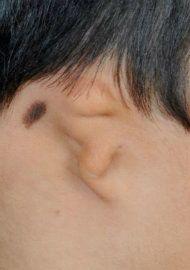
Lobular microtia with very small lobule
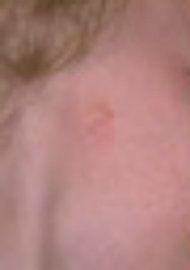
Anotia (complete absence of ear)

Dr. Thorne is the Editor-in-Chief and the author of several chapters in Grabb and Smith's PLASTIC SURGERY, 7th Edition.







